Setting up workspace
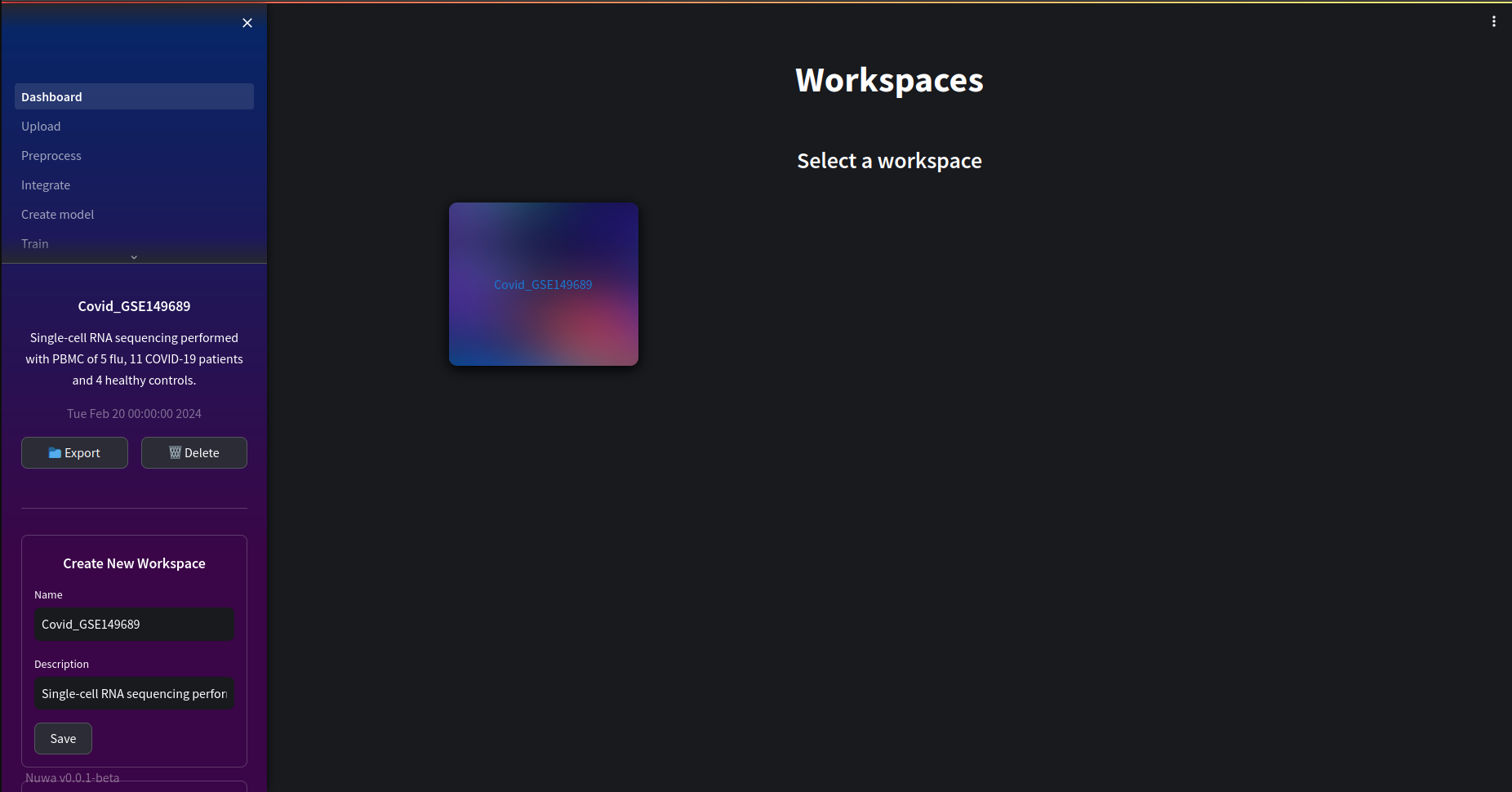
To start our analysis we first need to set up a workspace environment which will load in our datasets and create other files necessary for storing analysis results. Creating a workspace also allows us to store information relating to our investigation in a database. This includes file locations where results are stored, the different datasets used in the investigation and scripts which allow us to reproduce any analysis in the future.
Creating a workspace just requires a name and description of the investigation:
Next, in the upload page select the covid dataset. You may wish to learn more about the dataset at ncbi.
Obs and var
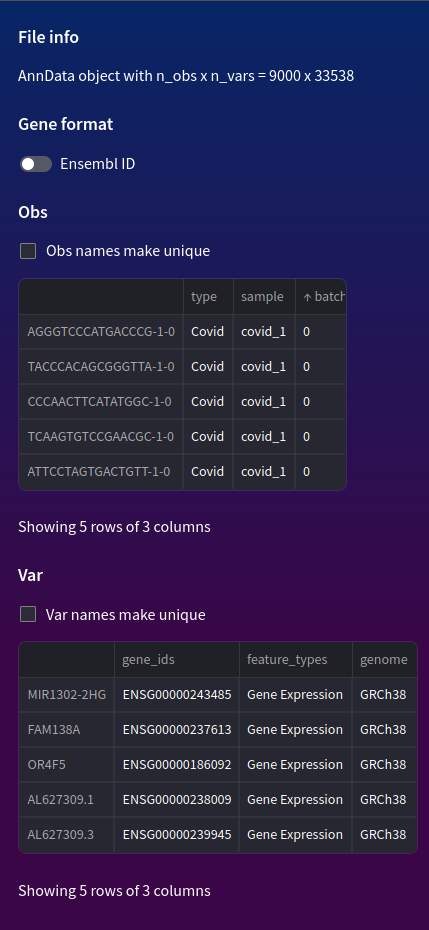
In the file info in the sidebar we can see there are 9000 obs and 33538 vars.
What are obs?
Obs (observations) include things like cell annotations (e.g. tumour or normal cell) or other information relating to the dataset.
In our case we have 3 obs columns:
- Type: Whether the patient has covid or not
- Sample: Identifier for sample donor
- Batch: Identifier for sample batch
What are vars?
Var (variables) refers to annotation of gene/feature metadata. This is a dataframe indexed by unique gene names (or other gene identifier such as ensembl ID).
In our case we have 3 columns:
- gene_ids: Ensembl ID of gene
- feature_types: what the type of data represents (i.e "gene_expression" to signify values are gene counts)
- genome: Reference genome which these genes were mapped to (in our case human reference genome GRCh38, or the 38th build of Genome Reference Consortium human)
To see more about the structure of Scanpy's anndata oject see here
Other useful functions
-
Gene format: the format of gene symbols (the way the .var dataframe is indexed) whether they are ensembl IDs or gene symbols. It is recommended not to change these unless necessary.
-
Obs/var make names unique: Make obs/var names unique
Other dataset sources
Generally you will be uploading your own datasets. Currently H5AD, H5, loom and mtx are supported. You can also import a dataset from the EBI expression atlas using an accession key.